Как уведомить MHRA о своем намерении провести клиническое исследование медицинских изделий.
Клинические исследования медицинских изделий
Как уведомить MHRA о своем намерении провести клиническое исследование медицинских изделий.
В рамках процесса получения маркировки UKCA, CE или CE UKNI для вашего медицинского изделия может потребоваться проведение клинического исследования. Вы должны уведомить MHRA о своих планах по проведению такого исследования не позднее чем за 60 дней до его начала.
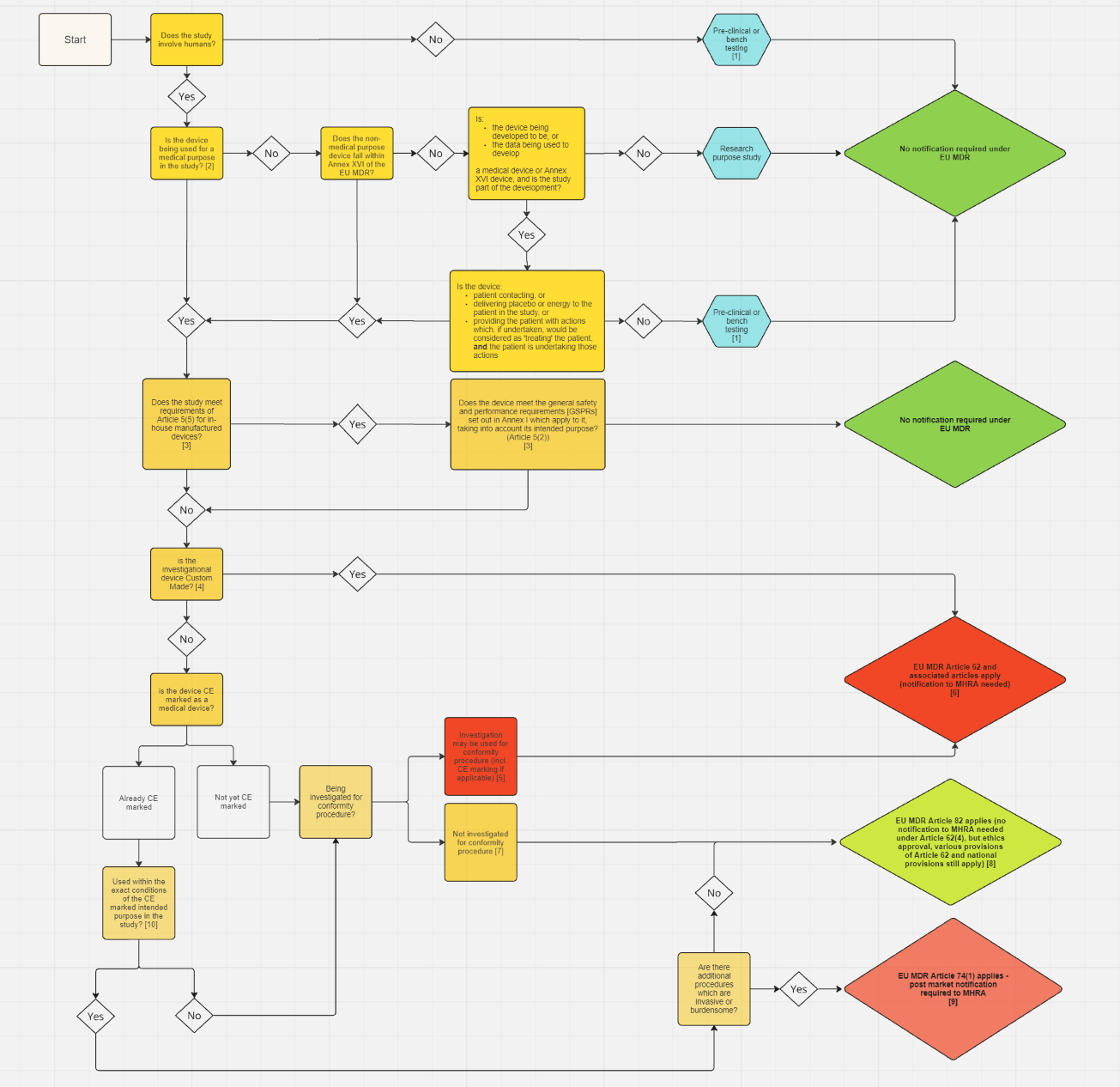
Требуется ли для моего исследования подача заявки на проведение клинического исследования?
Для исследований в Великобритании, пожалуйста, используйте эту блок-схему и прилагаемые к ней рекомендации , чтобы определить, нужно ли вам подавать заявку в MHRA. В августе 2025 года MHRA опубликовала обновленные версии этих документов. Пожалуйста, ознакомьтесь с последними доступными рекомендациями и убедитесь, что вы осведомлены об их содержании в отношении заявок на клинические исследования, поданных с этой даты.
Для обучения в Северной Ирландии или Великобритании и Северной Ирландии используйте блок-схему и прилагаемые рекомендации в разделе «Северная Ирландия» ниже .
Примечание: уведомление в MHRA не потребуется для медицинских изделий, имеющих маркировку UKCA, CE или CE UKNI, предназначенных для целей, находящихся на стадии исследования.
Заявки, поданные в соответствии с Приложением XVI к Регламенту (ЕС) 2017/745 (MDR) о медицинских изделиях, не предназначенных для медицинского использования, не принимаются в Великобритании.
Особые условия для медицинских учреждений
Вам не нужно уведомлять нас о проведении клинического исследования, если:
- Вы произвели медицинское изделие собственными силами для своих пациентов без цели вывести его на рынок.
Вам может потребоваться уведомить MHRA о проведении клинического исследования, если:
- Вы хотите предоставить другой организации медицинское изделие, которое до сих пор производилось внутри компании для пациентов, для получения данных, подтверждающих безопасность и эффективность коммерческого продукта.
Узнайте больше об исключении для медицинских учреждений в отношении медицинских изделий общего назначения, чтобы выяснить, распространяется ли это исключение на ваше исследование.
Пожалуйста, ознакомьтесь с блок-схемой и прилагаемыми инструкциями , чтобы определить, нужно ли вам подавать заявку в MHRA.
Связаться с нами
Запросы на проведение клинических исследований
По вопросам, касающимся заявок на проведение клинических исследований, обращайтесь по адресу info@mhra.gov.uk , указав в теме письма «запрос о клиническом исследовании».
Консультации по вопросам регулирования
Группа специалистов MHRA по клиническим исследованиям может предложить комплексную консультацию по вопросам регулирования медицинских изделий.
Мы не можем анализировать отдельные документы, но можем предоставить рекомендации по ориентированию в нормативно-правовой среде.
Ознакомьтесь с информацией о стоимости консультаций по вопросам регулирования.
Для запроса консультации по вопросам регулирования свяжитесь с руководителем отдела клинических исследований по адресу mark.grumbridge@mhra.gov.uk .
Сборы
См. раздел «Цены» на нашей странице .
Сбор в соответствии с Правилами обращения с медицинскими изделиями 2002 года:
(а) подлежит уплате с момента направления соответствующего уведомления государственному секретарю;
и
(b) должен прилагаться к этому уведомлению при его вручении.
Указанные на нашей странице тарифы сборы также покрывают расходы на подачу заявок на проведение исследований медицинских устройств в Северной Ирландии.
Оплата клинического исследования
Подробную информацию о том, как произвести оплату в MHRA, можно найти здесь .
Мы выставим счет на оплату, и чтобы помочь нам с этим, пожалуйста, как можно скорее предоставьте нам номер заказа на покупку и укажите, кто несет ответственность за оплату.
После получения счета и осуществления платежа, вы должны отправить подтверждение оплаты по электронной почте на адрес CI-applications@mhra.gov.uk и вашему назначенному специалисту по регулированию, указав номер ссылки MHRA (CI/XXXX/XXX).
Мы не будем предоставлять окончательное решение регулирующих органов (одобрение или возражение) по заявке до получения оплаты.
Упрощение платежей для малых и средних предприятий
- Для проведения клинических исследований малым и средним предприятиям предоставляются сервитуты. При подаче заявки необходимо предоставить письмо-разрешение от MHRA для малых и средних предприятий, чтобы сервитут был рассмотрен.
- После получения подтверждения заявки MHRA запросит 50% от суммы сбора, а оставшиеся 50% должны быть оплачены в течение шести месяцев после выставления первого счета.
- Если не будет оплачено оставшиеся 50% задолженности, то любое разрешение на проведение клинического исследования, выданное MHRA, будет аннулировано, что приведет к прекращению клинического исследования.
- В случае возражения или отзыва заявления, оставшаяся сумма сбора подлежит оплате.
- Для проведения встреч по вопросам регулирования медицинских устройств не предоставляется никаких послаблений.
- Для получения дополнительной информации см. раздел « Упрощение и отмена платежей для малых и средних предприятий».
Пилотный проект программы освобождения от платы за клинические исследования медицинских изделий для микро- и малых предприятий.
Программа освобождения от платы для микро- и малых предприятий, планирующих проводить клинические исследования с использованием инновационных медицинских устройств.
MHRA продлевает программу освобождения от платы за медицинские услуги на 2026-2027 годы, начиная с 20 апреля 2026 года.
В общей сложности десяти малым и микропредприятиям Великобритании, отвечающим установленным критериям, будет предоставлено освобождение от платы за подачу заявки на проведение клинических исследований.
Цель программы — поддержать доступ пациентов к инновационным устройствам путем тестирования влияния отмены регуляторных издержек на малые предприятия, где сборы могут выступать препятствием для роста или инноваций.
Подробности программы освобождения от платы за обучение
Продолжительность: Программа будет действовать с 20 апреля 2026 года по 5 апреля 2027 года или до тех пор, пока не будут предоставлены все льготы по оплате обучения.
Доступны льготы: Всего будет доступно десять льгот по оплате для устройств классов I, IIa, IIb, а теперь также и класса III, но не для активных имплантируемых устройств.
Результат: Для успешно прошедших отбор заявителей плата за подачу заявки на клиническое исследование будет полностью отменена.
Примечание:
- От каждого заявителя может быть одобрено только одно освобождение от требований.
- Мы принимаем заявки на исследования, включающие CTIMP, однако освобождение от платы распространяется только на часть заявки, касающуюся самого устройства, а полная плата за рассмотрение CTIMP по-прежнему должна быть внесена.
Условия получения освобождения от требований
Для участия необходимо выполнить следующие условия:
- Заявитель может продемонстрировать, что устройство соответствует следующим критериям «инновационного устройства»:
- Продукт представляет собой либо новый продукт, либо оригинальную модификацию существующих технологий;
- В Великобритании не существует решений, которые бы удовлетворяли тем же клиническим потребностям и имели бы соответствующее разрешение регулирующих органов;
- Устройство может быть масштабировано с конечной целью принести пользу системе здравоохранения и социального обеспечения, пациентам и/или поставщикам услуг по уходу.
- Заявитель должен подтвердить, что он проживает в Великобритании и получил статус микро- или малого предприятия от MHRA до подачи заявления на освобождение от требований.
См. определения для малых и микропредприятий .
Проверьте, соответствуете ли вы критериям для инновационных устройств.
Перед подачей официальной заявки через IRAS заявителям следует получить подтверждение от MHRA о том, что их устройство соответствует критериям инновационного устройства для получения разрешения на использование непродуманного устройства.
Для этого, пожалуйста, отправьте сопроводительное письмо по адресу CI-applications@mhra.gov.uk, содержащее:
- подробное описание устройства и
- как оно соответствует трем вышеупомянутым критериям инновационного устройства, и
- краткое изложение запланированного клинического исследования
Группа клинических исследований рассмотрит предоставленную информацию в течение 5 рабочих дней с момента получения и подтвердит следующее:
- соответствует ли исследование критериям инновационного устройства, и
- Есть ли возможность предоставления льгот по оплате сборов?
Процесс подачи заявки
- Заявители, планирующие подать заявку на проведение клинических исследований в MHRA, должны сначала проверить, соответствует ли их устройство критериям инновационных устройств для программы, проводимой MHRA. Инструкции по этому вопросу приведены в разделе выше.
- Если MHRA подтвердила соответствие критериям инновационного устройства, заявитель должен получить статус малого или микропредприятия от MHRA, следуя процедуре, описанной здесь .
- После того как MHRA подтвердит соответствие критериям и получение статуса малого или микропредприятия, заявитель должен подать заявку через IRAS .
Это следует сделать как можно скорее после обращения в MHRA, чтобы обеспечить дальнейшую доступность исключений. - MHRA проверит заявку в соответствии со стандартными процедурами, и, если она будет признана действительной, заявитель будет уведомлен о предоставлении освобождения от уплаты сбора.
Если заявка будет признана недействительной, заявитель будет уведомлен о причинах этого решения и получит возможность повторно подать заявку.
При повторном обращении для проверки заявитель может уточнить возможность получения освобождения от уплаты сбора, связавшись с CI-applications@mhra.gov.uk .
Дополнительная информация
Для получения информации о подаче заявки на получение статуса МСП см. раздел « Упрощение и отмена платежей для малых и средних предприятий» .
Если у вас возникнут дополнительные вопросы относительно пилотного проекта по освобождению от платы за обучение, отправьте электронное письмо по адресу CI-applications@mhra.gov.uk .
Для получения информации о ходе рассмотрения заявки на получение статуса малого или среднего предприятия, пожалуйста, свяжитесь с нами по адресу sales-invoices@mhra.gov.uk .
Начало работы над вашей заявкой
Как уведомить MHRA о проведении клинического исследования
Начните подачу заявки через IRAS . Заявки подаются в электронном виде с использованием Интегрированной системы подачи заявок на исследования (IRAS).
Руководство MHRA
Ознакомьтесь с рекомендациями по составлению заявки и руководством для производителей из Великобритании или руководством для производителей из Северной Ирландии при подготовке заявления на уведомление.
Узнайте, как MHRA утверждает клинические исследования, о которых нас уведомляют.
Ознакомьтесь с нашей информацией для клинических исследователей, чтобы узнать, что требуется от врачей, участвующих в исследовании.
Ознакомьтесь с рекомендациями по электродиагностике для проведения клинических исследований .
Ознакомьтесь с нашими рекомендациями по оценке биологической безопасности медицинских изделий, находящихся на стадии клинических исследований.
Ознакомьтесь со статистическими аспектами клинических исследований при представлении статистической информации, полученной в ходе вашего клинического исследования.
Ознакомьтесь с рекомендациями по применению принципов эргономики и проектирования удобства использования к медицинским изделиям, включая комбинированные лекарственно-медицинские препараты .
См. руководство по маркировке UKCA .
Контрольный список проверки
После получения вашей заявки на клиническое исследование медицинского изделия наши специалисты по регулированию проверят её соответствие контрольному списку для заявок на клинические исследования . Используйте этот контрольный список, чтобы подать действительную заявку.
Оценка
Данный раздел касается клинических исследований, проводимых исключительно в Великобритании. Информация об исследованиях, проводимых в Северной Ирландии, представлена в отдельном разделе ниже.
После получения и проверки ваших документов мы свяжемся с вами в течение 5 рабочих дней, чтобы подтвердить начало 60-дневного периода оценки, или сообщим о любых выявленных проблемах. В случае возникновения проблем, 60-дневный период оценки начнет отсчитываться с момента получения нами действительного ответа.
Первый день из 60 дней считается первым днем, следующим за датой принятия действительной заявки. Например, если заявка получена 24 августа, а эксперт подтверждает ее действительность 28 августа, отсчет времени начинается с 29 августа.
В ходе оценки эксперты оценят безопасность и работоспособность вашего устройства, а также составят план проводимого клинического исследования. Мы свяжемся с вами, если нам потребуется дополнительная информация. Крайне важно связаться с нами как можно скорее, если вам потребуется уточнение.
Если имеются основания для возражений, мы, по возможности, организуем телеконференцию для лучшего понимания ситуации и поиска решения в течение 60-дневного срока оценки.
Мы вышлем вам письмо к 60-му дню с решением («возражение» или «отсутствие возражений») о том, можете ли вы провести предложенное клиническое исследование.
Заявки на экспериментальный лекарственный препарат и медицинское изделие — параллельное рассмотрение.
Данный порядок подачи заявок распространяется на клинические исследования, которые одновременно являются клиническим испытанием исследуемого лекарственного препарата (CTIMP) и клиническим исследованием (CI) медицинского изделия.
В подобных ситуациях исследования следует представлять в MHRA в виде заявки на «параллельное рассмотрение» (ранее известной как комбинированное исследование исследуемого лекарственного препарата и исследуемого медицинского изделия (IMP+Device)).
Пожалуйста, ознакомьтесь с последними рекомендациями, доступными здесь:
Данный механизм призван преодолеть разрыв между двумя отдельными сводами законов, сроками их принятия и функциональностью системы.
В долгосрочной перспективе подразделение MHRA по клиническим исследованиям и испытаниям намерено максимально согласовать правила и процессы, чтобы упростить процедуру подачи заявок на исследования, находящиеся на стадии параллельного рассмотрения.
Также ознакомьтесь с последними рекомендациями по клиническим испытаниям: Лекарственные препараты: центр клинических испытаний — GOV.UK
Поправки
Данный раздел касается клинических исследований, проведенных исключительно в Великобритании. Информация об исследованиях, проведенных в Северной Ирландии, представлена в отдельном разделе ниже.
После получения от нас письма об отсутствии возражений, вы должны уведомить нас обо всех предлагаемых изменениях в расследовании. Вы должны дождаться, пока мы пришлем вам еще одно письмо об отсутствии возражений, прежде чем вносить изменения.
Вы обязаны сообщить нам о любых изменениях, внесенных в:
- исследуемое устройство
- документация исследования, включая план клинического исследования.
- следователей или следственные учреждения
- изменения, запрошенные комитетом по этике
Если вы не сообщите нам о предлагаемых поправках, вы можете быть привлечены к уголовной ответственности.
Начните подачу поправки на сайте HRA IRAS.
Обратитесь к разделу о внесении изменений в руководстве пользователя IRAS .
При уведомлении MHRA об изменениях, укажите следующее:
- Сопроводительное письмо с указанием регистрационного номера MHRA для клинического исследования (например, CI/2023/XXXX).
- Таблица с кратким описанием каждого предлагаемого изменения и обоснованием каждого изменения.
- Отмеченные красным цветом (показывающие вносимые изменения) и чистые копии всей измененной документации исследования
- Подписанное производителем или от его имени заявление о том, что предлагаемые изменения не приведут к предсказуемому увеличению риска для пациента, пользователя или третьей стороны.
Если вы не будете следовать приведенным выше инструкциям, ваша заявка будет возвращена вам.
Отправляйте поправки по электронной почте на адрес CI-amendments@mhra.gov.uk , за исключением случаев, когда файлы слишком большие; в противном случае свяжитесь с нами по тому же адресу электронной почты, чтобы запросить ссылку для загрузки документов. Мы будем обрабатывать только поправки, отправленные на этот почтовый ящик. Не отправляйте поправки отдельным лицам или на другие почтовые ящики.
Оплата за внесение поправок
В соответствии с новыми тарифами, введенными в июле 2025 года, плата за внесение каких-либо изменений в результаты клинического обследования не взимается.
Требования к отчетности
Сообщение о серьезных нежелательных явлениях (СНЯ).
Этот раздел касается клинических исследований, проводимых на площадках в Великобритании, а также клинических исследований, проводимых на площадках как в Великобритании, так и в Северной Ирландии. Информацию об исследованиях, проводимых только на площадках в Северной Ирландии (площадки в Великобритании отсутствуют), см. в отдельном разделе ниже .
В соответствии с правилами Великобритании и директивой MEDDEV 2.7/3, все подлежащие регистрации события должны быть полностью зарегистрированы и сообщены в MHRA. Это включает в себя все серьезные нежелательные явления, независимо от того, была ли признана причинно-следственная связь между устройством и событием, подлежащим регистрации (согласно MEDDEV 2.7/3), происходящие в третьих странах, где клиническое исследование проводится в рамках того же плана клинического исследования. Эти данные могут быть предоставлены с использованием либо таблицы отчетности о серьезных нежелательных явлениях MEDDEV 2.7/3, либо таблицы отчетности о серьезных нежелательных явлениях MDCG 2020-10/2, при условии включения необходимой информации.
Пожалуйста, заполните форму отчета о нежелательных явлениях (SAE) и отправьте ее через новый портал MORE , прикрепив к ней заполненную таблицу.
Подробную информацию о регистрации на портале MORE можно найти здесь .
В документе MEDDEV 2.7/3 также содержатся дополнительные рекомендации по отчетности о клинических исследованиях в Великобритании.
Квартальные сводные отчеты (КСО)
В качестве условия одобрения MHRA для проведения клинического исследования, помимо сообщения об отдельных серьезных нежелательных явлениях, как указано выше, вы должны ежеквартально направлять нам сводные отчеты, содержащие обновленную информацию о последнем общем профиле безопасности исследования.
Если исследование проводится только в Великобритании, мы ожидаем, что первый отчет QSR будет представлен через квартал после того, как первый участник пройдет лечение.
Для исследований, в которых также участвуют центры в ЕС и по всему миру, пожалуйста, предоставьте отчет о качестве исследования (QSR) через квартал после одобрения исследования MHRA в Великобритании. В отчете должно быть подробно описано общее состояние безопасности для ВСЕХ исследовательских центров.
Для составления таких сводных отчетов используйте . Этот шаблон предназначен только для медицинских изделий, но его также следует использовать для отчетности, связанной с медицинскими изделиями, в рамках любых объединенных исследований. Не включайте подробную информацию о каких-либо исследуемых лекарственных препаратах (ИЛП).
Отправляйте свои квартальные сводные отчеты напрямую через портал MORE.
Дополнительные рекомендации по подаче заявок на регистрацию предприятий общественного питания (SAE и QSR) в систему MORE можно найти в руководстве пользователя по подаче заявок в MORE .
Исследование отклонений
Спонсоры обязаны уведомлять MHRA обо всех отклонениях (только в отношении исследовательских центров в Великобритании), как только им станет о них известно. Необходимо указать подробности о характере отклонения, времени его возникновения, месте его возникновения, а также о любых предлагаемых корректирующих и превентивных мерах.
При составлении отчетов об отклонениях используйте следующий шаблон Excel для отслеживания отклонений от протокола MHRA и храните его как «живой» документ, чтобы можно было добавлять новые отклонения. Это позволит как спонсору, так и MHRA иметь полный обзор при каждом представлении отчета.
Отправьте заполненную электронную таблицу по электронной почте на адрес info@mhra.gov.uk .
Досрочное прекращение или временная приостановка клинического исследования
Спонсоры обязаны уведомить нас о досрочном прекращении клинического исследования и предоставить обоснование досрочного прекращения, как указано в Положениях 16(11) и Разделе 29(10) Регламента Великобритании о медицинских изделиях 2002 года.
Отправьте копию окончательного письменного отчета о клиническом исследовании устройства, подпадающего под действие Регламента Великобритании о медицинских изделиях 2002 года (Регламенты 16(10) и 29(9)), по адресу CI-applications@mhra.gov.uk .
Спонсоры также должны уведомить нас о временной приостановке клинического исследования.
Отправляйте уведомления по адресу info@mhra.gov.uk .
Отчеты по итогам исследования
Отправьте отчет об окончании исследования по электронной почте на адрес CI-applications@mhra.gov.uk .
Северная Ирландия
Протокол по Северной Ирландии требует, чтобы Северная Ирландия продолжала соответствовать правилам ЕС в отношении медицинских изделий после 1 января 2021 года. Таким образом, Регламент о медицинских изделиях (ЕС) 2017/745 (MDR) и Регламент о медицинских изделиях для диагностики in vitro (ЕС) 2017/746 (IVDR) будут применяться в Северной Ирландии с 26 мая 2021 года и 26 мая 2022 года соответственно, в соответствии с графиком внедрения, установленным ЕС.
Все клинические исследования, требующие подачи заявки в MHRA (которые проводятся на территории Северной Ирландии), должны быть представлены в MHRA в соответствии с требованиями Регламента ЕС о медицинских изделиях 2017/745.
Примечание: данная единая заявка также будет распространяться на любые площадки, предлагаемые для проведения клинического исследования в Великобритании, а также на площадки в Северной Ирландии.
У нас есть рекомендации для производителей в Северной Ирландии .
Требуется ли для моего исследования подача заявки на проведение клинического исследования?
Для клинических исследований, проводимых на территории Северной Ирландии, используйте эту блок-схему и прилагаемые к ней рекомендации , чтобы определить, нужно ли вам подавать заявку в MHRA (Агентство по регулированию лекарственных средств и изделий медицинского назначения Великобритании).
Обратите внимание: MHRA недавно опубликовала обновленную блок-схему для Великобритании и сопутствующие руководства (см. ссылки вверху этой страницы). В рамках этого рабочего пакета блок-схема для Северной Ирландии и сопутствующие руководящие документы также находятся на стадии пересмотра и могут быть обновлены в установленном порядке. Пожалуйста, регулярно проверяйте эту страницу, чтобы быть в курсе последних рекомендаций по этой теме.
В следующих разделах представлены рекомендации по подаче таких заявок, а также требования к внесению изменений и проведению постмаркетинговых исследований, касающихся объектов в Северной Ирландии.
Оценка применения когнитивных имплантатов
После получения и проверки ваших документов мы свяжемся с вами в течение 10 календарных дней, чтобы подтвердить действительность заявления и начало рассмотрения, или сообщим о любых проблемах. В случае возникновения проблем мы подтвердим это в письменной форме и предоставим 10-дневный срок для ответа. Рассмотрение не начнется до получения нами действительного ответа. Если после получения ответа мы по-прежнему сочтем заявление недействительным или 10-дневный срок истек, мы подтвердим это в течение 5 календарных дней.
Первый день оценки MHRA считается датой подтверждения получения нами действительной заявки. В ходе оценки эксперты оценят безопасность и эффективность вашего устройства, а также план клинического исследования, которое будет проведено.
Ваша заявка будет рассмотрена (и может быть запрошена дополнительная информация) в соответствии с процедурами и сроками, описанными в руководстве MHRA для производителей (ссылка находится вверху этой страницы). Крайне важно, чтобы вы связались с нами как можно скорее, если вам потребуется уточнение. Если есть основания для отказа в выдаче разрешения, по возможности мы организуем телеконференцию для лучшего понимания ситуации и поиска решения в течение периода рассмотрения.
Мы вышлем вам письмо к последнему дню (или раньше) с решением о том, сможете ли вы провести предложенное клиническое исследование.
Поправки/изменения
После получения от нас разрешения на проведение клинического исследования в Северной Ирландии, вы должны уведомить нас обо всех предлагаемых изменениях в исследовании до внесения каких-либо изменений. Необходимо дождаться, пока мы вышлем вам еще одно письмо с разрешением, прежде чем вносить существенные изменения. В зависимости от того, будем ли мы консультироваться с экспертами, мы вышлем вам наше решение в течение 38 или 45 календарных дней.
Вы обязаны сообщить нам обо всех изменениях, но только те, которые будут сочтены существенными, потребуют разрешения от MHRA.
Существенные изменения включают в себя:
- изменения в исследуемом медицинском устройстве
- изменения в дизайне или методологии клинического исследования, а также в исходной информации.
- изменения в процедурах, проводимых участниками
- изменения в оценке соотношения риска и пользы для исследования
- существенные изменения в документации исследования, такие как план клинического исследования, брошюра исследователя, информационные листы для участников, формы согласия, письма врачам общей практики или другим медицинским работникам, информационные листы для родственников или лиц, осуществляющих уход.
- назначение нового главного следователя
- Включение нового места проведения испытаний (не указанного в первоначальной заявке).
- назначение нового главного исследователя на площадке проведения клинических испытаний
- Плановое возобновление исследования после временной остановки, если были внесены изменения в устройство или исследование.
- изменение определения окончания исследования
- продление исследования сверх срока, указанного в заявлении.
- любые другие существенные изменения в протоколе
- изменения, запрошенные комитетом по этике
В случае несущественных изменений вам достаточно уведомить нас, чтобы обеспечить актуальность наших записей. Если после рассмотрения предлагаемого несущественного изменения мы сочтем его существенным, мы сообщим об этом спонсору, и предлагаемое изменение не должно быть внесено до получения разрешения MHRA.
К несущественным изменениям относятся такие изменения, которые вряд ли окажут существенное влияние на безопасность, здоровье или права участников исследования, а также на достоверность и надежность клинических данных, полученных в ходе исследования, и включают в себя:
- смена спонсора(ов) или законного представителя спонсора
- изменение условий страхования или возмещения убытков, связанных с исследованием.
- Незначительные изменения в протоколе или другой документации исследования, например, исправление опечаток, обновление контактных данных, незначительные уточнения.
- изменения в исследовательской группе главного исследователя
- изменения в составе исследовательской группы на отдельных площадках проведения испытаний (за исключением назначения нового главного исследователя)
- изменения в порядке финансирования
- Незначительные изменения в документации, используемой исследовательской группой для регистрации данных исследования.
- изменения в логистических процедурах хранения или транспортировки образцов
Требования к отчетности
Сообщение о серьезных нежелательных явлениях (СНЯ) в соответствии с Регламентом (ЕС) 2017/745
Этот раздел касается клинических исследований, проводимых в Северной Ирландии (но не в Великобритании). Информацию об исследованиях, проводимых в Великобритании, или об исследованиях, проводимых как в Великобритании, так и в Северной Ирландии, см. в отдельном разделе выше .
К данным исследованиям применяются требования к отчетности, установленные Регламентом (ЕС) 2017/745.
Спонсор обязан в полном объеме регистрировать всю следующую информацию:
- любое нежелательное явление, указанное в плане клинического исследования как критически важное для оценки результатов этого клинического исследования;
- любое серьезное неблагоприятное событие;
- любой недостаток устройства, который мог бы привести к серьезному неблагоприятному событию, если бы не были приняты соответствующие меры, не было бы проведено вмешательство или обстоятельства сложились бы менее благоприятно;
- любые новые данные, касающиеся любого события, упомянутого в пунктах (а) – (с).
Спонсор обязан незамедлительно сообщить в MHRA обо всем следующем:
- любое серьезное нежелательное явление, имеющее причинно-следственную связь с исследуемым устройством, компаратором или процедурой исследования, или в случаях, когда такая причинно-следственная связь является разумно возможной;
- любой недостаток устройства, который мог бы привести к серьезному неблагоприятному событию, если бы не были приняты соответствующие меры, не было бы проведено вмешательство или обстоятельства сложились бы менее благоприятно;
- любые новые данные, касающиеся любого события, упомянутого в пунктах (а) и (б).
Отчеты можно предоставлять, используя таблицу отчетности SAE MDCG 2020-10/2.
Пожалуйста, заполните форму отчета SAE и приложите к ней заполненную таблицу на портале MORE .
Подробную информацию о регистрации на портале MORE можно найти здесь .
В документе MDCG 2020-10 также содержатся дополнительные рекомендации по отчетности о результатах клинических исследований в Северной Ирландии.
По всем вопросам обращайтесь к разделу выше на этой странице с инструкциями.
Досрочное прекращение или временная остановка клинических исследований
Спонсоры обязаны уведомить MHRA о временной приостановке или досрочном прекращении клинического исследования и предоставить обоснование в течение 15 дней, или 24 часов, если решение было принято по соображениям безопасности. Отчет о клиническом исследовании и его краткое изложение должны быть предоставлены в течение 3 месяцев для таких исследований, которые временно приостановлены или досрочно прекращены.
Постмаркетинговые исследования
Вы обязаны уведомить нас обо всех клинических исследованиях, проведенных в Северной Ирландии с использованием устройств, имеющих маркировку CE, которые также включают процедуры, выходящие за рамки обычных условий использования устройства, и которые являются инвазивными или обременительными.
Уведомление необходимо подать не позднее чем за 30 дней до начала исследования.
При предоставлении нам информации о проведении этих клинических исследований нам необходима следующая информация:
- форма заявления
- брошюра следователя
- план или протокол клинического исследования
- подписанное заявление
- Мнение REC
- подтверждение наличия страхового полиса или возмещения убытков субъектам
- ПИС и ИК
- соглашение, обеспечивающее защиту и конфиденциальность персональных данных.
- подробная информация о хранящейся технической документации (анализ рисков, протоколы испытаний) доступна.
Подайте заявку в электронном виде, используя интегрированную систему подачи заявок на исследовательские проекты (IRAS) .
Медицинские изделия: клинические исследования и исследования эффективности в Северной Ирландии
Информация об изменениях в проведении некоторых клинических исследований и исследований эффективности в Северной Ирландии.
Для проведения некоторых клинических исследований и исследований эффективности в Северной Ирландии спонсоры должны быть зарегистрированы либо в Северной Ирландии, либо в ЕС, либо иметь законного представителя, зарегистрированного в Северной Ирландии или ЕС.
Действующие правила (Регламент о медицинских изделиях (Регламент 2017/745) (MDR) и Регламент о диагностике in vitro (Регламент 2017/746) (IVDR)) позволяют Великобритании не применять эти правила.
Это требование применяется, если исследование проводится только на территории организации, а не в ЕС. Таким образом, MHRA отменяет требование о том, чтобы спонсор или его законный представитель клинического исследования или исследования эффективности был зарегистрирован в Северной Ирландии или ЕС, при условии соблюдения всех следующих условий:
- Расследование или исследование также должно проводиться как в Северной Ирландии, так и в Великобритании.
- Расследование или исследование не должно проводиться в государстве-члене ЕС.
- Спонсор должен быть либо зарегистрирован в Великобритании, либо иметь письменное соглашение с юридическим представителем в Великобритании, который несет ответственность за обеспечение соблюдения спонсором обязательств, предусмотренных Регламентом о медицинских изделиях (MDR) или Регламентом о инфекционных заболеваниях (IVDR).
- Спонсор должен назначить контактное лицо в Северной Ирландии для проведения клинического исследования или исследования эффективности, которое будет адресатом для всей переписки со спонсором, предусмотренной в Регламенте о медицинских изделиях (MDR) или Регламенте о внутривенном введении (IVDR).
- Любое общение с этим контактным лицом считается общением со спонсором.
Все остальные требования, предусмотренные Регламентами о медицинских изделиях (MDR) и Индукционными регламентами по инфекционному контролю (IVDR), остаются в силе. Данное руководство вступает в силу немедленно.
Для получения рекомендаций по представлению результатов исследований эффективности в MHRA в соответствии с IVDR, пожалуйста, ознакомьтесь с руководством IRAS по IVD и Северной Ирландии .
Для получения дополнительной информации обращайтесь в MHRA по адресу info@mhra.gov.uk .
Одобрение Управления по исследованиям в области здравоохранения (HRA) и Управления по исследованиям в области здравоохранения и ухода в Уэльсе (HCRW).
Одобрение HRA и HCRW распространяется на все проектные исследования, проводимые в Национальной службе здравоохранения Англии и Уэльса. Оно объединяет оценку соблюдения нормативных требований и законодательства, проводимую специально назначенными сотрудниками HRA и HCRW, с независимым заключением Комитета по этике исследований (REC), предоставляемым Службой этики исследований Великобритании.
Это заменяет необходимость проведения каждой участвующей организацией в Англии и Уэльсе местных проверок соблюдения законодательства и связанных с этим вопросов. Информацию о том, как подготовить и подать заявку на утверждение HRA и HCRW, можно найти на веб-сайте HRA .
Более подробная информация
Более подробную информацию о классификациях см. в документе MEDDEV 2.4/1, содержащем рекомендации по классификации .
Дополнительные рекомендации по клиническим испытаниям лекарственных препаратов (КИПЛ) см. на сайте: Клинические испытания и исследования — GOV.UK.
Если у вас возникнут какие-либо вопросы перед отправкой уведомления, напишите по адресу info@mhra.gov.uk .
Последнее обновление: 6 июля 2026 Показать все обновления

{kind=link}
{kind=link}
{kind=link}